
Case contribution: Dr. Radhiana Hassan
Clinical:
- A 39 years old female
- Initial presentation 4 years ago with right paraesthesia and left upper and lower limb weakness.
- MRI Brain showed multiages non traumatic intracranial haemorrhages and haematomyelia with upper cervical cord oedema.
- Claimed symptoms resolved after 6 months. Plan to repeat mri but pt defaulted subsequently.
- Then presented again early this year with gradual worsening of right paraesthesia and left sided weakness.
- Neurology power over the left upper and lower limb 4/5, hyperreflexia and sensation intact.
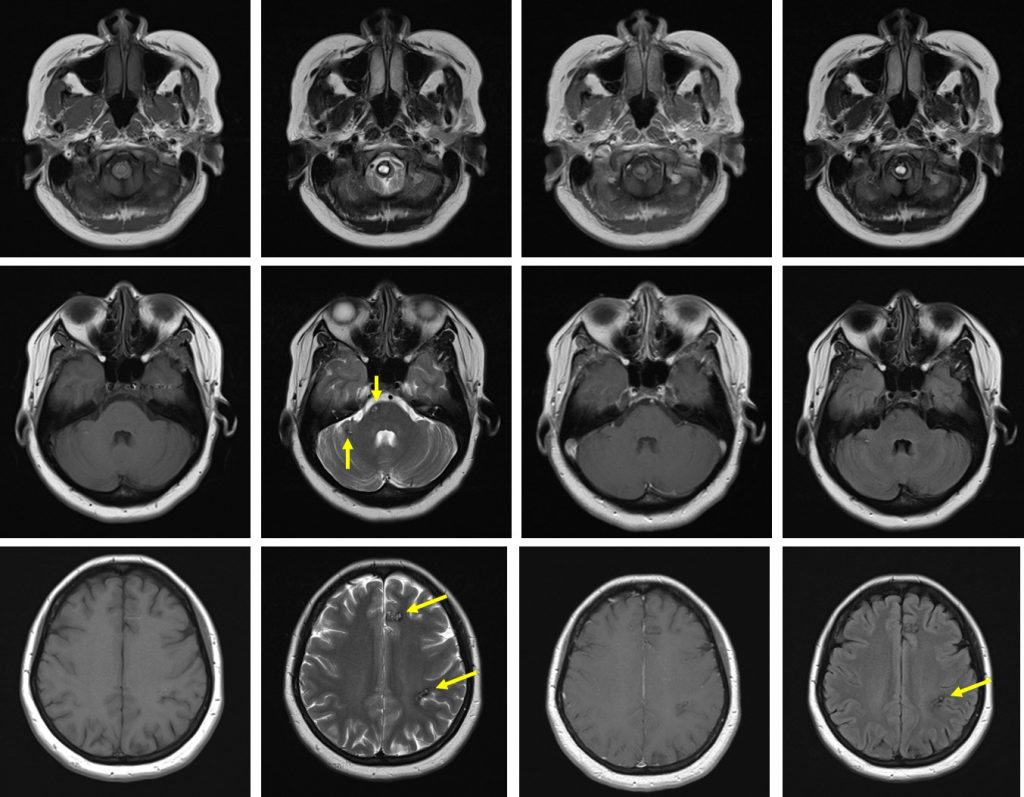
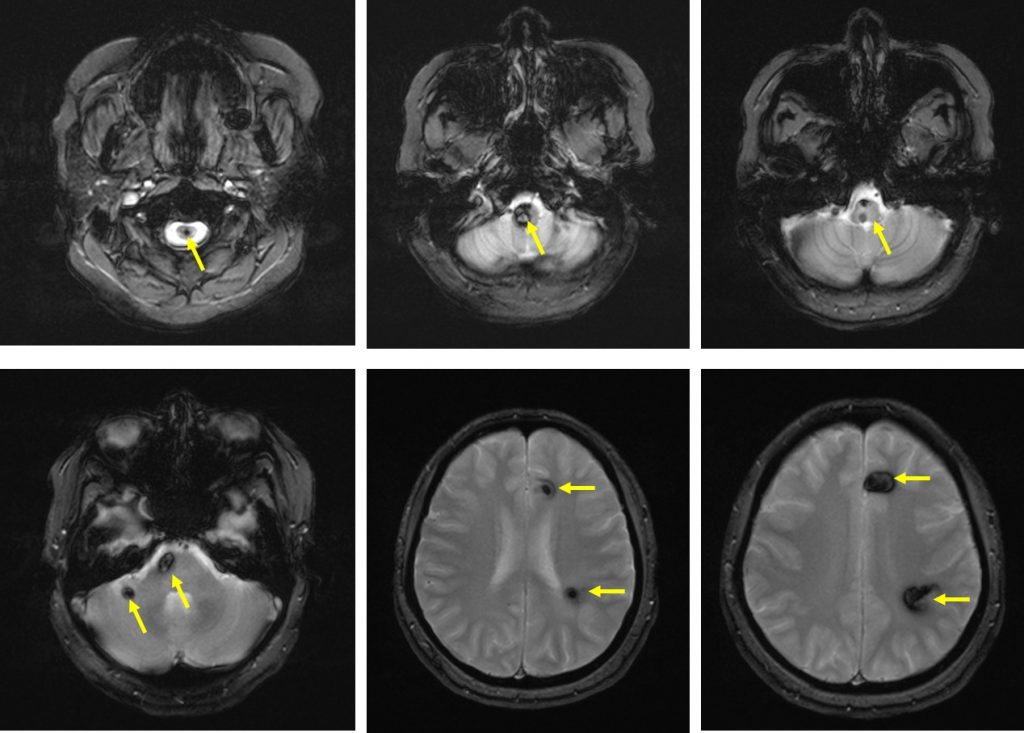
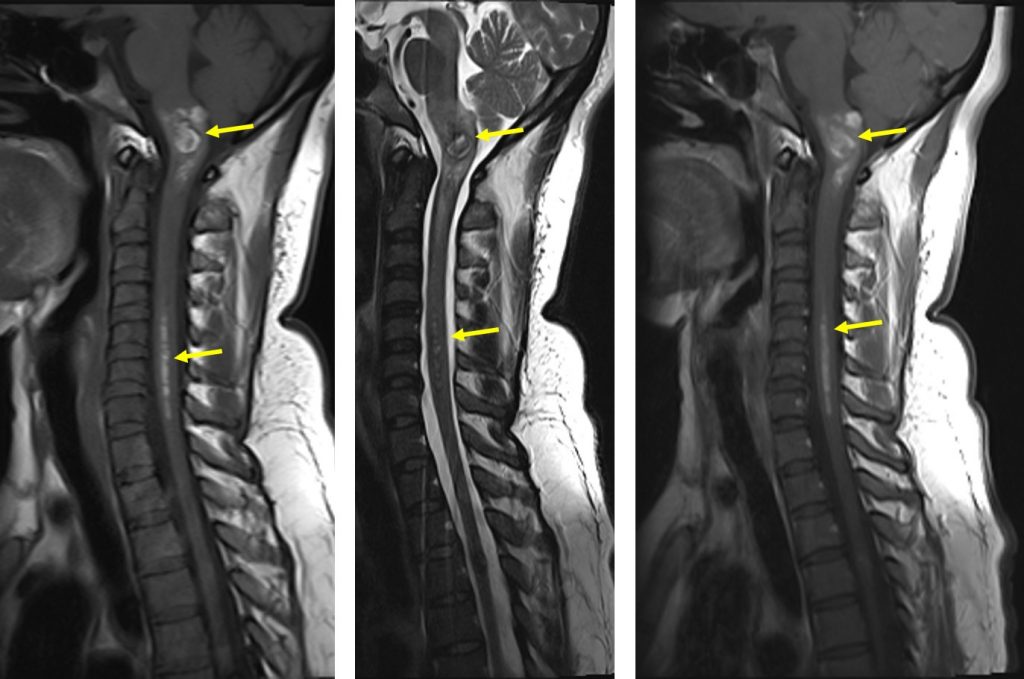
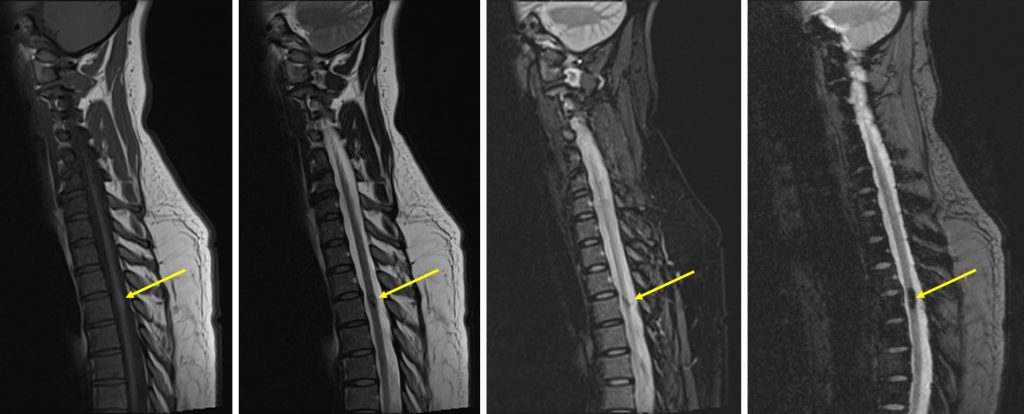
MRI findings:
- MRI of brain and spine shows multiple lesions in the brain parenchyma and spinal cord (yellow arrows)
- These lesions are not well seen on T1, heterogenous on T2, not enhanced post contrast with blooming artifact on Hemo sequences
- These lesions are of varying ages, no surrounding oedema is seen
- The larger lesions show characteristic popcorn/mulberry appearance on T2-weighted images
Diagnosis: Multiple cavernomas/familial cavernomas
Discussion:
- Cerebral cavernous malformations are multiple mulberry-like distended caverns of dilated thin-walled capillaries without the normal intervening brain parenchymal architecture. It is often surrounded by hemosiderin representing remote oozing due to abnormal capillaries
- CCM is the second most common vascular malformations of CNS after DVA.
- It is commonly seen at supratentorial cerebri and spinal cord involvement is rare.
- The mean age of presentation is around 37 years, however it can present at any age. It has been reported to be higher incidence in certain ethnic (Hispanic population).
- There are 3 protein-encoding genes (CCM1, CCM2 and CCM3) known to cause familial CCM. Mutation in these genes is sufficient for causing the disease.
- Familial CCM is defined as the presence of multiple CCM (typically five or more). Diagnostic criteria (at least one of the following)
- Presence of multiple CCM (5 or more)
- The occurrence of CCM in at least 2 members of a family
- The presence of mutation in one of three genes causing FCCM
- In familial CCM, about 20-50% of cases remain asymptomatic and are incidentally found during imaging.
- The recurrent microhemorrhages in CCM that result in nearby deposition of hemosiderin and gliosis and inflammation around the lesion are believed to be the cause of seizures in CCM patients.
- An annual ICH risk of 1.5% to 4.6% among the patients and ICH risk of 0.1% to 1.4% for each lesion per year. An overall ICH risk of 15.8% has been estimated for CCM patients with the yearly risk of recurring ICH decreasing with every passing year. This is an important factor to consider best treatment option for patients.
- Diagnosis of CCMs can be a challenge as compared with other vascular diseases.
- They are not detectable on cerebral angiography as catheter angiography can only identify potential abnormal venous flow associated with CCMs.
- Similarly, small lesions may not be detected on computed tomography scan.
- Hence, magnetic resonance imaging (MRI) is the modality of choice for evaluating CCMs.
- MRI sequences should include typical T1- and T2-weighted sequences including T2 FLAIR and T2* sequences, preferably including susceptibility-weighted imaging (SWI) or similar susceptibility-sensitive sequence. T2*-weighted gradient recalled echo sequences are more sensitive for smaller CCMs than conventional T2 sequences
- It is recommended that the MRI of suspected CCMs in brain and/or spinal cord should include SWI of the brain to confirm the lesion and evaluate for DVAs.
- If imaging studies reveal a solitary CCM associated with a DVA, there is more likelihood of a diagnosis of sporadic CCM, though, FCCM can rarely be associated with DVAs as well.